")
Rétinoblastome héréditaire : estimer le risque et conseiller la famille

Related content
Lorsqu’un diagnostic de rétinoblastome héréditaire ne peut être confirmé par analyse génétique, il est possible d’estimer le risque encouru par les autres membres de la famille et de l’utiliser pour planifier des examens oculaires de suivi.
Les laboratoires de génétique peuvent détecter beaucoup de mutations différentes dans le gène du rétinoblastome et les tests génétiques sont utilisés pour déterminer si un individu, et les membres de sa famille, présente(nt) un risque significatif de développer un rétinoblastome.
Dans les pays à faible ou moyen revenu, toutefois, beaucoup de centres de traitement n’ont pas la possibilité de réaliser une analyse génétique. Dans ce cas, il revient au clinicien d’évaluer le risque que son jeune patient présente un rétinoblastome héréditaire, en se basant sur le tableau clinique et les antécédents familiaux :
- Le rétinoblastome héréditaire se développe à un plus jeune âge (âge médian de 15 mois).
- Les tumeurs sont généralement bilatérales et multifocales, mais elles peuvent parfois être unilatérales.
- D’autres membres de la famille sont affectés ; ils présentent soit un rétinoblastome soit, plus rarement, une seconde tumeur.
- Le père ou la mère, ou un autre parent proche, a subi une énucléation durant son enfance.
- L’examen révèle une tumeur bénigne de la rétine (rétinome) chez les parents ou les frères et soeurs (page 30).
Lorsqu’un enfant est atteint d’un rétinoblastome de forme héréditaire, ses frères et soeurs (et ses futurs enfants) sont susceptibles de développer la maladie. Ils doivent donc subir un examen complet de la rétine à intervalles réguliers, ce afin de détecter et traiter toute tumeur potentielle le plus tôt possible.
Si un enfant présente un rétinoblastome unilatéral, il faut toujours examiner l’autre oeil avec soin jusqu’à l’âge de cinq ans, pour détecter et traiter tout signe de rétinoblastome bilatéral.
Estimation du risque de rétinoblastome héréditaire
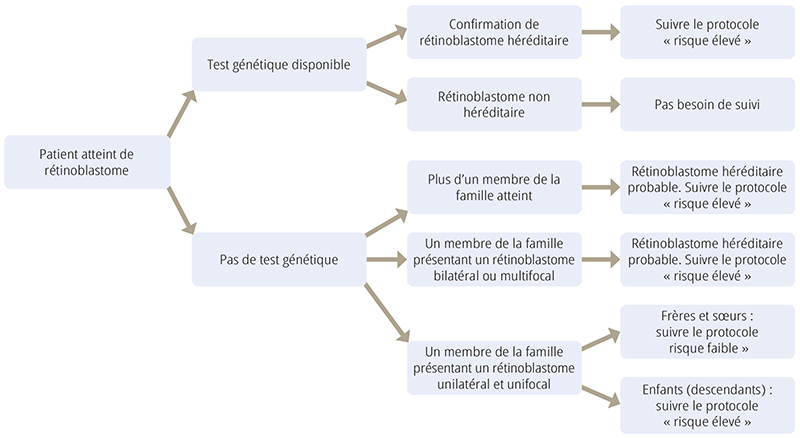
Pour étayer la prise de décision en l’absence de test génétique, il est possible d’utiliser l’algorithme présenté dans la Figure 1 (page 32) pour estimer le risque encouru par les autres membres de la famille lorsqu’un enfant est atteint de rétinoblastome. Cet algorithme a été développé après avoir passé en revue les résultats des enfants ayant subi une analyse génétique dans notre service.
Le Tableau 1 présente une estimation du risque que les membres de la famille d’un patient atteint de rétinoblastome (frère ou soeur, enfant, ou parent plus éloigné) développent eux aussi une tumeur de la rétine. Les deux questions les plus importantes sont : « Existet- il au moins un autre membre de la famille atteint de rétinoblastome ? » et « Le rétinoblastome est-il multifocal, bilatéral ou unilatéral ? ». Les catégories sont regroupées par niveau de risque : un risque supérieur à 1 % est considéré comme risque élevé et un risque inférieur ou égal à 1 % est considéré comme risque faible.
Servez-vous du Tableau 1 ; basez-vous sur le lien de parenté avec le patient et le tableau clinique pour déterminer s’il faut mettre en oeuvre un protocole de dépistage à « risque élevé » (orange) ou à « risque faible » (vert). Le Tableau 2 à la page 32 décrit ces protocoles de dépistage, soit quand et à quelle fréquence les membres de la famille devront subir un examen détaillé de la rétine pour dépister à un stade précoce toute tumeur potentielle.
Tableau 1 Estimation du risque de rétinoblastome chez les frères et soeurs et les futurs enfants d’un enfant atteint de rétinoblastome
| Plus d’un membre de la famille affecté | L’enfant est atteint de rétinoblastome bilatéral ou multifocal | L’enfant est atteint de rétinoblastome unilatéral et unifocal | |
|---|---|---|---|
| Frères et soeurs de l’enfant atteint de Rb* | 50 % | 5 % | 1 % |
| Futurs enfants de l’enfant atteint de Rb* | 50 % | 50 % | 5 –10 % |
| Parents plus éloignés de l’enfant atteint de Rb* | Étudier l’arbre généalogique, peuvent être à risque | Pas de risque accru | Pas à risque |
Il est important de parler avec les parents et les personnes qui s’occupent de l’enfant, afin de les informer du risque encouru par leur enfant et du fait que ses frères et soeurs, ainsi que ses futurs enfants, risquent eux aussi de développer un rétinoblastome. Soulignez que des examens oculaires à intervalles réguliers (Tableau 2) permettront le diagnostic précoce d’une éventuelle tumeur et favoriseront donc le succès du traitement. Soyez à l’écoute des inquiétudes des parents. Assurezvous qu’ils savent exactement où se rendre pour faire examiner les autres membres de la famille et consacrez suffisamment de temps à la discussion pour répondre à toute question éventuelle.
Tableau 2 Protocoles de dépistage (« risque élevé » et « risque faible ») pour les futurs enfants ou les frères et soeurs des enfants atteints de rétinoblastome
| Âge des frères et soeurs ou de l’enfant | Risque élevé | Risque faible |
|---|---|---|
| 2 semaines | Au plus tard à 2 semaines | |
| 4 semaines | Au plus tard à 4 semaines | |
| Jusqu’à 6 mois | Toutes les 4 semaines | Toutes les 6 semaines |
| 6 à 12 mois | Toutes les 4 à 6 semaines | À 9 mois et à 12 mois |
| 1 à 2 ans | Tous les 2 mois jusqu’à 18 mois
Puis à 21 mois et à 24 mois |
À 16 mois et à 22 mois |
| 2 à 3 ans | Tous les 4 mois | Tous les 6 mois |
| Après 3 ans | Arrêter, sauf si antécédents familiaux de déclenchement tardif de la maladie | Arrêter |
Secondes tumeurs
Les personnes atteintes de rétinoblastome héréditaire sont également à risque de développer d’autres tumeurs au cours de leur vie. Ces deuxièmes cancers sont le plus souvent un ostéosarcome, un sarcome des tissus mous ou un mélanome. Le risque augmente si l’enfant a été traité par radiothérapie externe. Les secondes tumeurs se développent le plus souvent entre 10 ans et 50 ans et peuvent apparaître n’importe où dans l’organisme. Il n’existe pas de dépistage efficace de ces deuxièmes tumeurs, donc il est important que le patient et son équipe soignante aient conscience qu’elles peuvent survenir. Il est important que le patient se protège du soleil et examine régulièrement sa peau pour repérer un mélanome éventuel.
Note : si un enfant présente un rétinoblastome unilatéral, il faut toujours examiner l’autre oeil avec soin, afin de détecter et traiter tout signe de rétinoblastome bilatéral.