")
Prise en charge du rétinoblastome à extension extraoculaire

Related content
Chez les enfants présentant une extension extraoculaire du rétinoblastome, le taux de survie peut être amélioré en associant chimiothérapie, chirurgie et radiothérapie.
Dans les pays à haut revenu, le taux de survie des enfants atteints de rétinoblastome s’est amélioré au fil des ans : il est passé de 5 % à plus de 95 %. Cette amélioration est le résultat d’un diagnostic et d’une prise en charge précoces par des équipes spécialisées dans le rétinoblastome. Toutefois, dans les pays à bas ou moyen revenu, le taux de survie reste bas, avec un taux de mortalité de 39 % en Asie et de 70 % sur le continent africain1. Le taux de mortalité élevé dans les pays à faible ou moyen revenu est principalement dû au délai entre l’apparition des symptômes et le début du traitement. Ce délai s’explique par un ensemble de facteurs, notamment un manque d’accès aux services de santé, de mauvaises conditions socio-économiques et une éducation insuffisante ; ceci fait que les patients consultent quand la maladie est à un stade avancé. Il faut ajouter à ces facteurs une mauvaise observance du traitement, notamment le refus de l’énucléation (un traitement pouvant potentiellement sauver la vie de l’enfant) pour des raisons culturelles.
Dans les pays à revenu élevé, le premier signe de rétinoblastome vu en consultation est souvent une leucocorie ou reflet blanc dans la pupille ; dans les pays à faible ou moyen revenu, on voit plutôt une buphtalmie ou une exophtalmie avec extension extraoculaire de la massetumorale2. On estimait autrefois que seulement 9 % des patients présentant un rétinoblastome à extension orbitaire (stade 3 de la maladie, voir page 34) survivaient plus de deux ans après leur diagnostic. Toutefois, des études récentes suggèrent que si l’on combine chimiothérapie, chirurgie et radiothérapie, le taux de survie à cinq ans des patients présentant le stade 3 de la maladie (rétinoblastome avec extension orbitaire) est supérieur à 50 %, bien que le pronostic demeure extrêmement mauvais pour les patients présentant le stade 4 de la maladie avec métastases systémiques ou cérebrales3.
Prise en charge recommandée en cas d’extension extraoculaire du rétinoblastome
Évaluation de l’étendue de la maladie
Effectuez un examen général, puis examinez les deux yeux avec soin, de préférence sous anesthésie. Dans la mesure où la moelle osseuse et le liquide céphalorachidien sont des sites métastatiques potentiels, il est recommandé de pratiquer une ponction de moelle osseuse et une analyse du liquide céphalorachidien avant de commencer le traitement. S’il y a augmentation de la taille des ganglions locaux, vous pouvez pratiquer une cytoponction ganglionnaire afin de déterminer s’il y a envahissement des ganglions lymphatiques. Il est recommandé de réaliser un scanner ou une IRM de l’orbite, afin de déterminer l’étendue de la maladie.
Traitement
Le protocole de traitement détaillé dans cet article est celui que nous utilisons dans notre service depuis l’année 2000. La prise en charge comporte plusieurs cycles de chimiothérapie systémique à forte dose, suivis par une intervention chirurgicale (énucléation ou exentération oculaire), puis une radiothérapie orbitaire externe. Si l’extension tumorale extraoculaire est évidente, y compris en cas d’envahissement extrascléral ou d’envahissement du nerf optique, le traitement de première intention doit être une chimiothérapie systémique à forte dose toutes les trois semaines. Il faut éviter de réaliser en première intention une énucléation ou une exentération orbitaire et d’abord attendre qu’il y ait régression tumorale, afin de maximiser la réussite de l’intervention chirurgicale et faire en sorte qu’elle ne laisse pas de résidu tumoral. Différents types de chimiothérapie ont été évoqués dans la littérature scientifique. Nous utilisons actuellement une association de vincristine, d’étoposide et de carboplatine, et nous trouvons cette chimiothérapie efficace (voir aussi Tableau 1 et Figure 1).
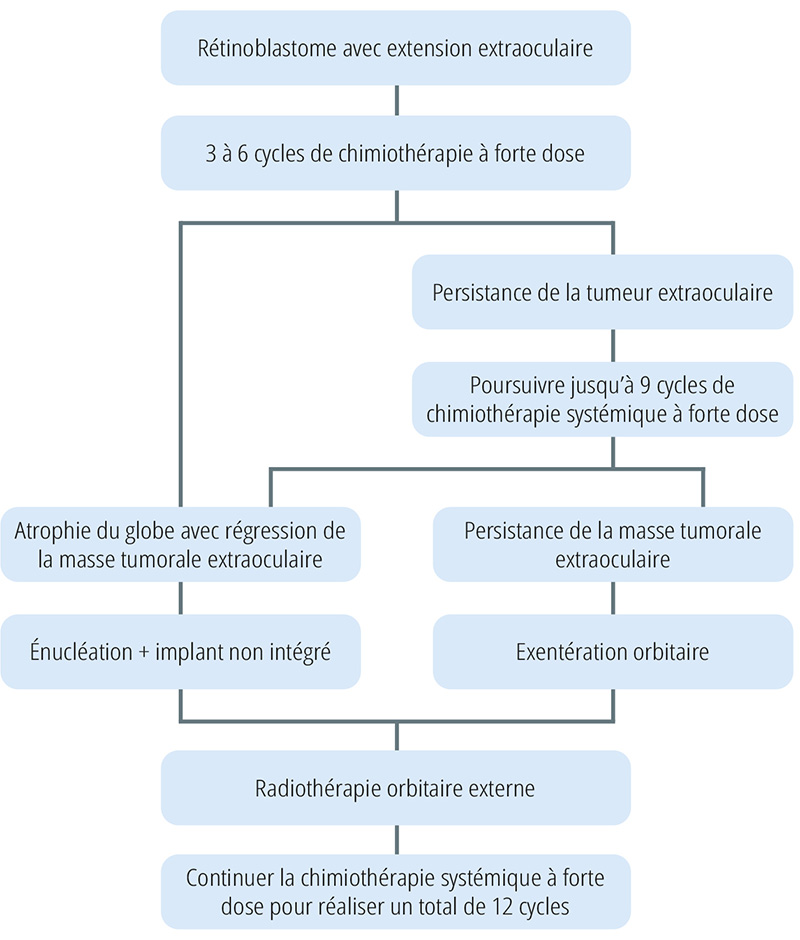
Tableau 1 Chimiothérapie systémique à forte dose pour extension extraoculaire du rétinoblastome
| Médicament | Posologie | |
|---|---|---|
| Enfants < 3 ans | Enfants > 3 ans | |
| Vincristine | 0,05 mg/kg | 1,5 mg/m2 de surface corporelle (S. C.) |
| Étoposide | 10 à 12 mg/kg | 200 mg/m2 de S. C. |
| Carboplatine | 28 mg/kg | 560 mg/m2 de S. C. |
| Schéma thérapeutique de chaque cycle | Médicaments administrés | |
| Jour 1 | Vincristide + Étoposide + Carboplatine | Vincristide + Étoposide + Carboplatine |
| Jour 2 | Étoposide | Étoposide |
La chimiothérapie à forte dose est poursuivie jusqu’à ce que l’on observe une régression de la masse tumorale extraoculaire. Après six cycles de chimiothérapie en moyenne, on obtient une régression complète de la masse tumorale extraoculaire dans 95 % des cas, ce qui permet d’éviter d’avoir à réaliser une exentération orbitaire (Figure 2). On peut réaliser à nouveau un scanner ou une IRM de l’orbite pour déterminer s’il y a régression ou persistance de la tumeur extraoculaire.
Figure 2 Traitement d’une patiente présentant un rétinoblastome à extension extraoculaire
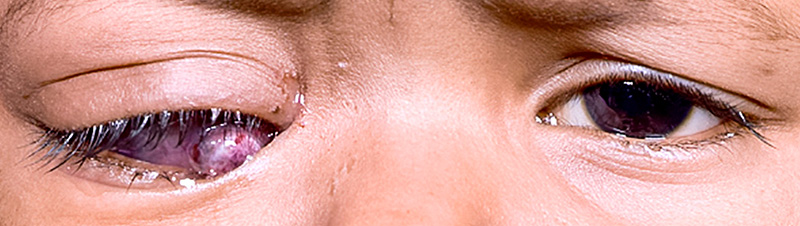
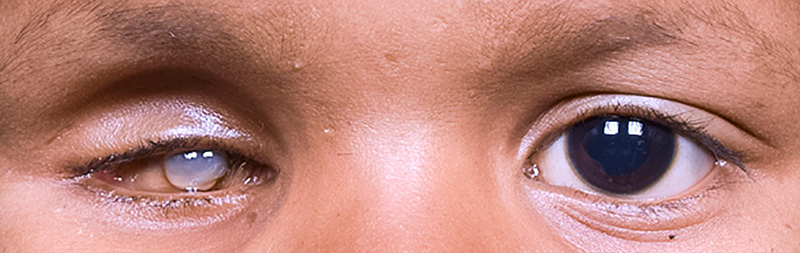

Lorsque l’on a obtenu une régression de la masse tumorale extraoculaire, on peut réaliser en seconde intention une énucléation avec placement d’un implant. Lorsqu’il y a toujours un résidu tumoral extraoculaire en dépit d’un maximum de neuf cycles de chimiothérapie, nous recommandons alors une exentération orbitaire en deuxième intention.
Six semaines après l’intervention chirurgicale, nous administrons une radiothérapie orbitaire externe (45 à 50 Gy). Le champ d’irradiation doit inclure les ganglions voisins si le patient présentait un envahissement des ganglions lymphatiques voisins lors de la première consultation.
Après la radiothérapie, nous continuons la chimiothérapie à forte dose jusqu’à ce que nous ayons réalisé 12 cycles de chimiothérapie au total. Chez les patients présentant le stade 4 de la maladie lors de la première consultation, nous recommandons également d’administrer en plus une chimiothérapie par voie intrathécale.
Nous avons analysé les résultats obtenus avec ce protocole thérapeutique dans notre service sur 20 patients présentant le stade 3 de la maladie et ayant bien suivi le traitement : 17 d’entre eux ont survécu et se portaient bien après une durée de suivi médiane de 77 mois et trois patients sont morts en dépit d’une bonne observance thérapeutique3. Tous les patients n’ayant pas bien suivi le traitement ont fini par mourir des suites de la maladie. Tous les patients qui s’étaient présentés en consultation avec le stade 4 de la maladie sont morts en dépit d’un traitement vigoureux et multimodal.
Pour résumer, un diagnostic précoce et une prise en charge appropriée du rétinoblastome sont essentiels pour augmenter les chances de survie du patient et les chances de sauver le globe oculaire et la vision du patient. Lorsqu’un patient consulte tardivement et présente une extension extraoculaire du rétinoblastome, mais sans métastases cérébrales ou systémiques, alors un traitement multimodal dans un centre spécialisé augmente les chances de survie. L’observance du traitement a une influence très importante sur les chances de survie.
Références
1 Kivelä T. The epidemiological challenge of the most frequent eye cancer: retinoblastoma, an issue of birth and death. Br J Ophthalmol 2009;93(9):1129-31.
2 Ali AA, et al. Clinical presentation and outcome of retinoblastoma among children treated at the National Cancer Institute (NCI) in Gezira, Sudan: a single Institution experience. Ophthalmic Genet 2011;32(2):122-5.
3 Kaliki S, et al. Clinical presentation and outcomes of stage III or stage IV retinoblastoma in 80 Asian Indian patients. J Pediatr Ophthalmol Strabismus 2017;54(3):177-184.