")
Back of the eye glossary
Related content
Diabetic retinopathy (DR)
Diabetic retinopathy can be subdivided into two basic forms:
1. Non-proliferative
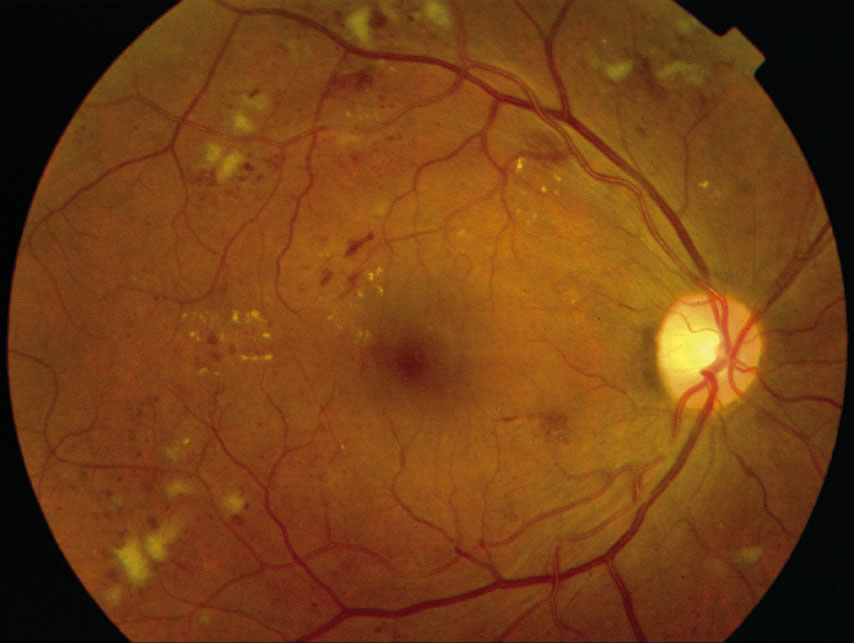
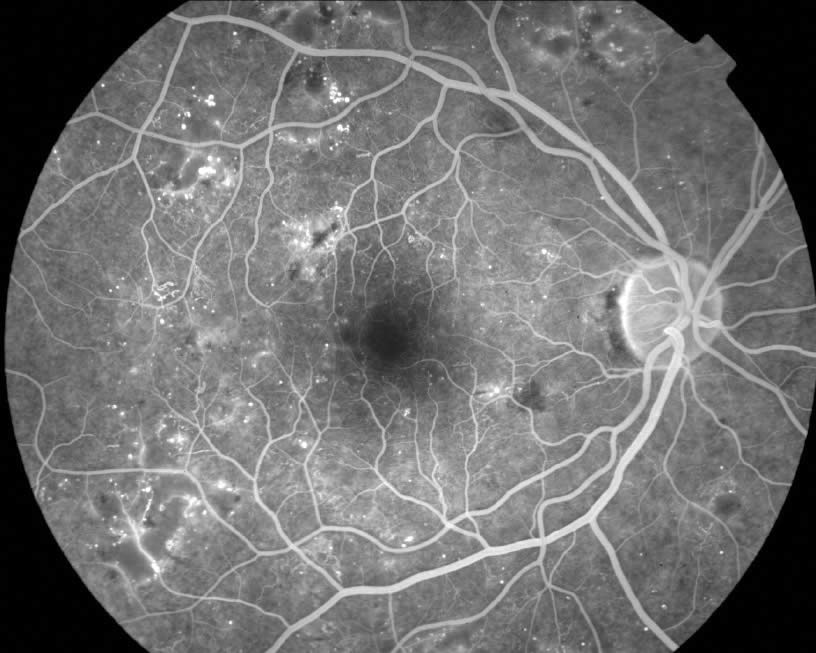
Non-proliferative disease can be identified by a number of clinical findings: microaneurysms, ‘dot and blot’ haemorrhages, cotton wool spots. Clinically significant macular oedema (CSME) is defined as:
- Retinal thickening within 500 microns of the centre of the fovea
- Hard exudation within 500 microns of the centre of the fovea if associated with retinal thickening
- Retinal thickening of one disc area, any part of which is located within one disc diameter (1,500 microns) from the centre of the fovea.
Eyes with CSME benefit from focal/grid laser photocoagulation to the macula.
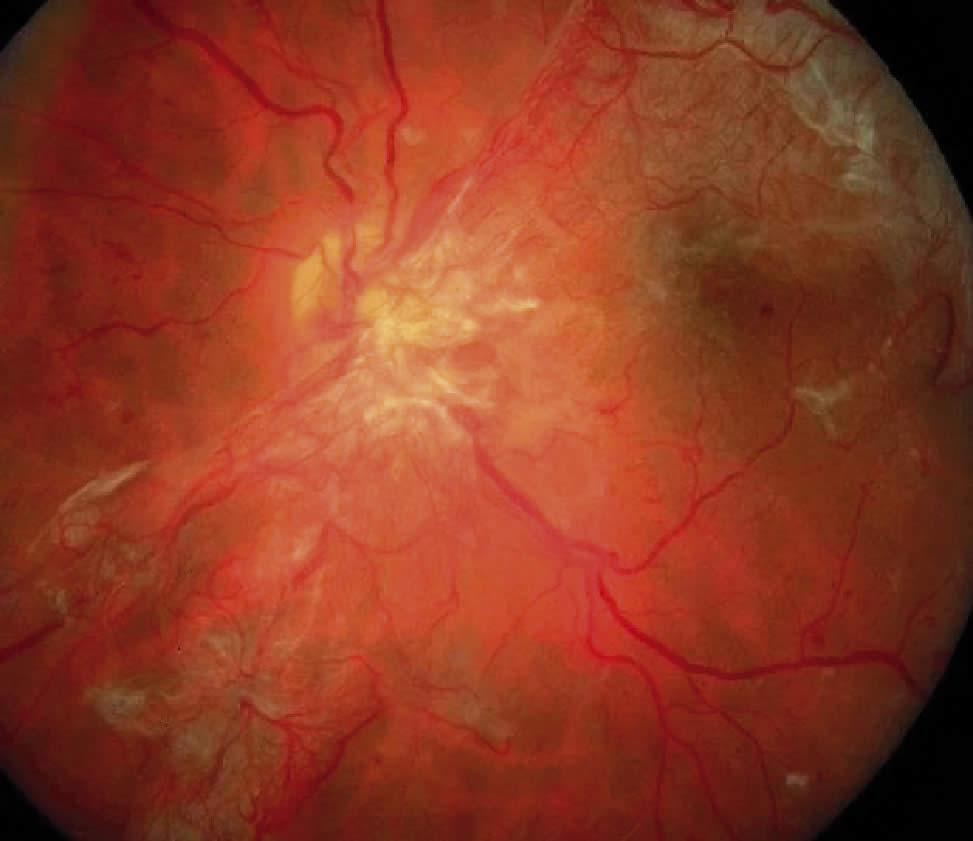
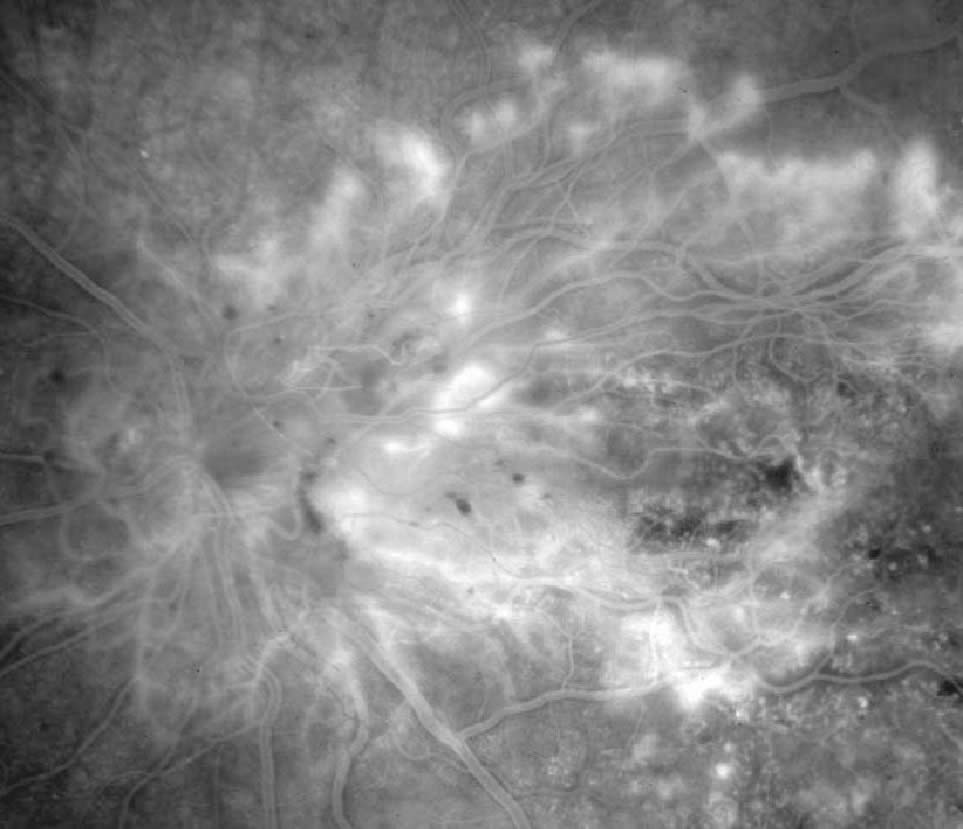
2. Proliferative
In proliferative diabetic retinopathy, the eyes demonstrate, singularly or in combination, neovascularisation of the disc (NVD), neovascularisation of the retina (neovascularisation elsewhere NVE), or neovascularisation of the iris (NVI) capillaries and veins. High-risk characteristics include:
- NVD greater than or equal to one fourth to one third of a disc area (one quarter of a disc area in eyes with large optic discs and one third of a disc area in eyes with small optic discs)
- NVD of any size associated with preretinal or vitreous bleeding
- NVE at least 0.5 disc area and associated with preretinal or vitreous bleeding. Eyes of high-risk characterisitics benefit from panretinal laser photocoagulation.
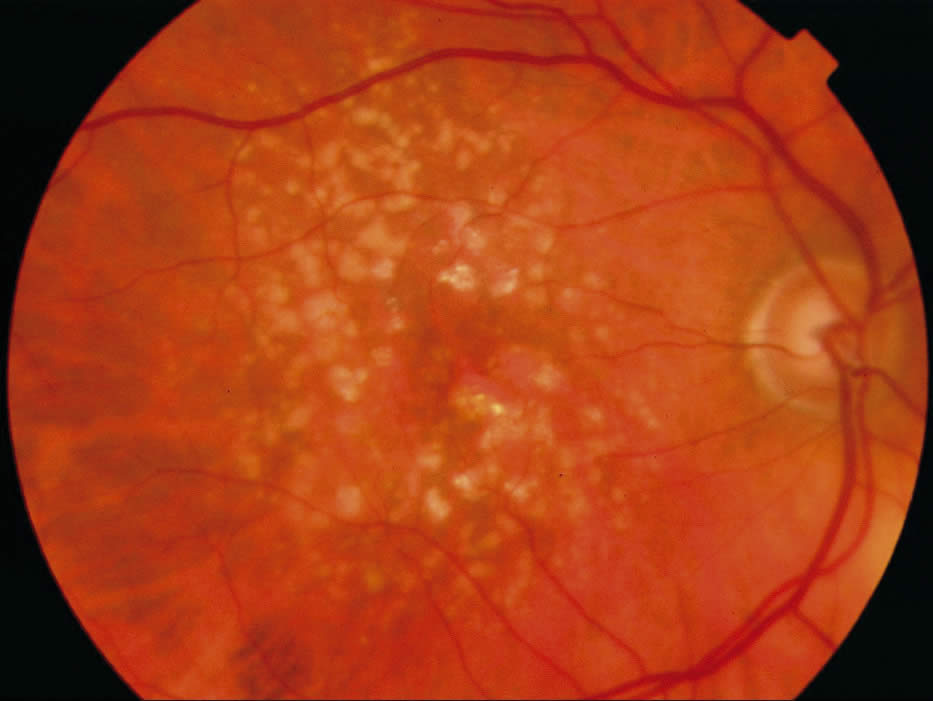
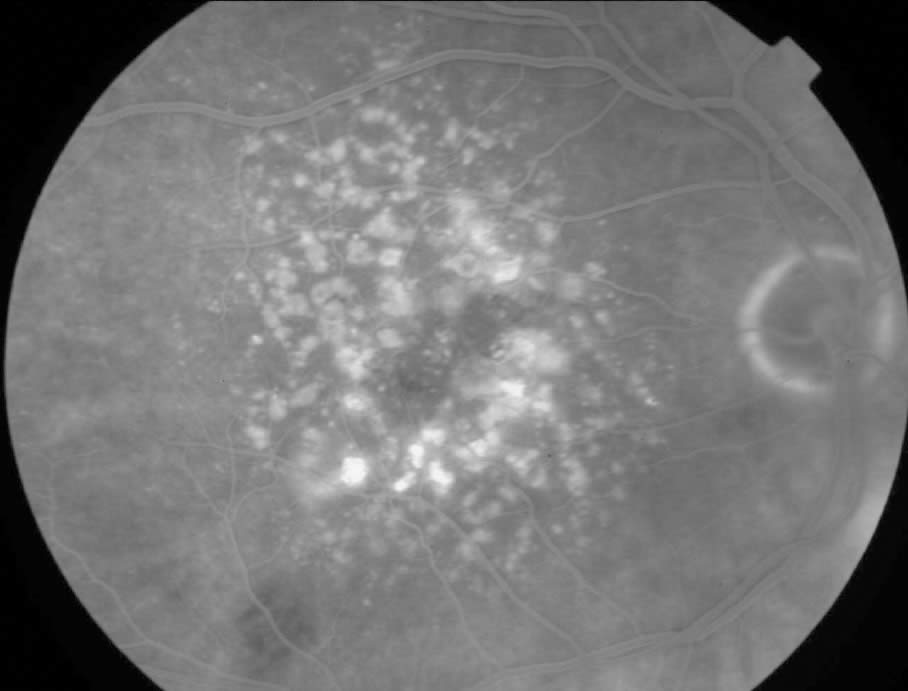
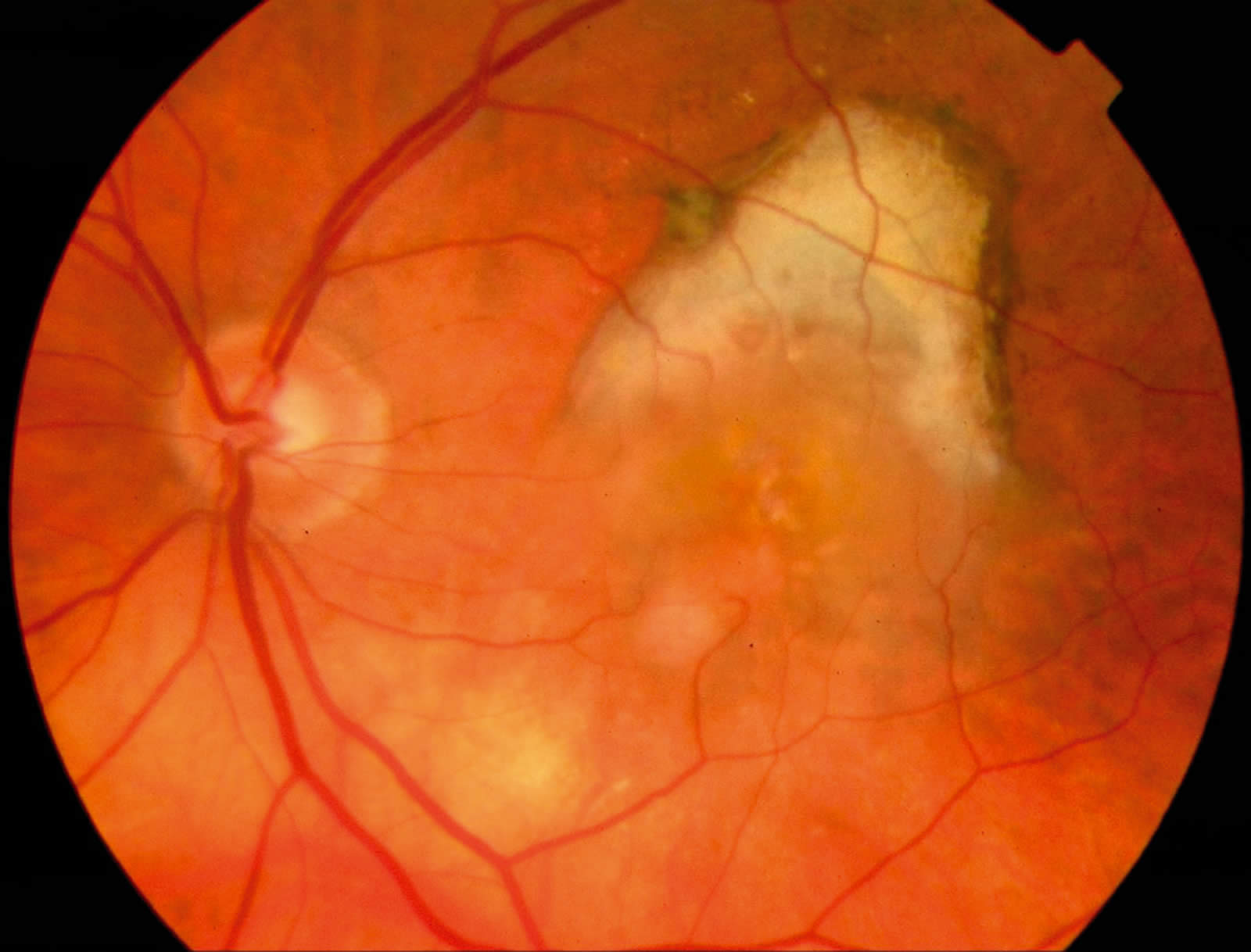
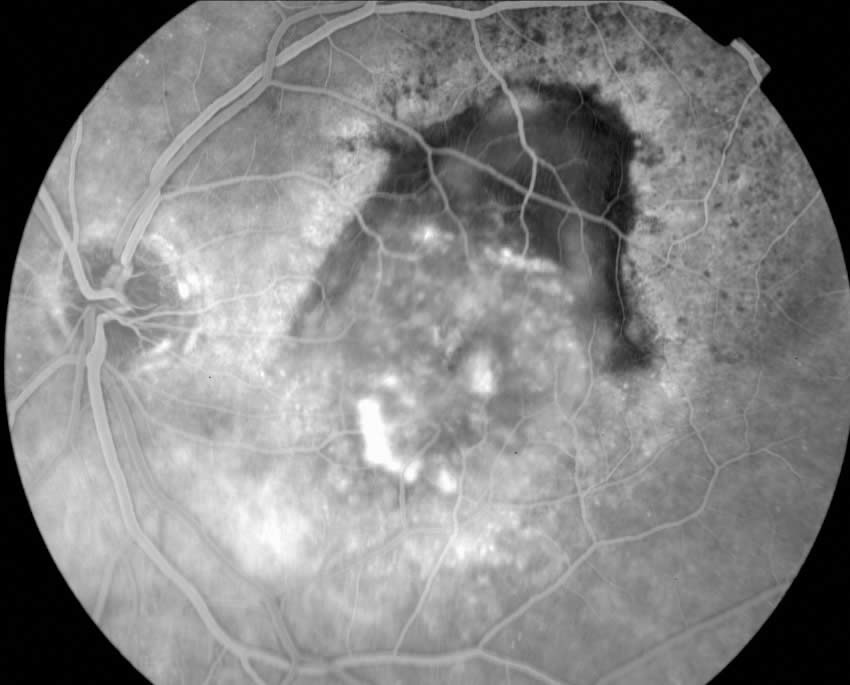
Age-related macular degeneration (AMD)
Age-related macular degeneration (AMD) is a progressive deterioration of Bruch’s membrane, retinal pigment epithelial, choriocapillaris, and outer retina in the macular area. There are two variants:
1. ‘Dry Type’:
Drusen and associated retinal pigment epithelial changes (atrophy and clumping). The majority of these eyes have moderate visual disturbance. Extensive or ‘geographic’ atrophy of the retinal pigment epithelium, however, can result in marked visual acuity loss.
2. ‘Wet or Exudative Type’:
Choroidal neovascularisation (or ‘membrane’) with associated fluid, lipid exudate, and haemorrhage under either the retinal pigment epithelium or neurosensory retina. This typically causes moderate to severe loss of central vision. The natural history is poor, often leading to subretinal fibrosis and scarring. This type can be further divided into the ‘classic’ membrane and the ‘occult’ membrane using fluorescein angiography.
Retinopathy of prematurity (ROP)
The condition was initially referred to as retrolental fibroplasia. There are five stages in classification:
Stage 1 is defined as a thin structure within the plane of the retina that separates vascularised from avascular retina.
Stage 2 represents an elevated ridge that has extended beyond the plane of the retina.
Stage 3: there is extraretinal fibrovascular proliferation or neovascularisation at the ridge.
Stage 4: there is a partial traction-like retinal detachment.
Stage 5 is defined as a total retinal detachment in an open or closed funnel configuration. The term ‘plus disease’ denotes significantly dilated and tortuous retinal vessels in the posterior pole. It indicates extensive vascular incompetence, and can be associated with vitreous haze, iris vessel engorgement, and poor pupillary dilation. ‘Plus disease’ is a poor prognostic sign in ROP.
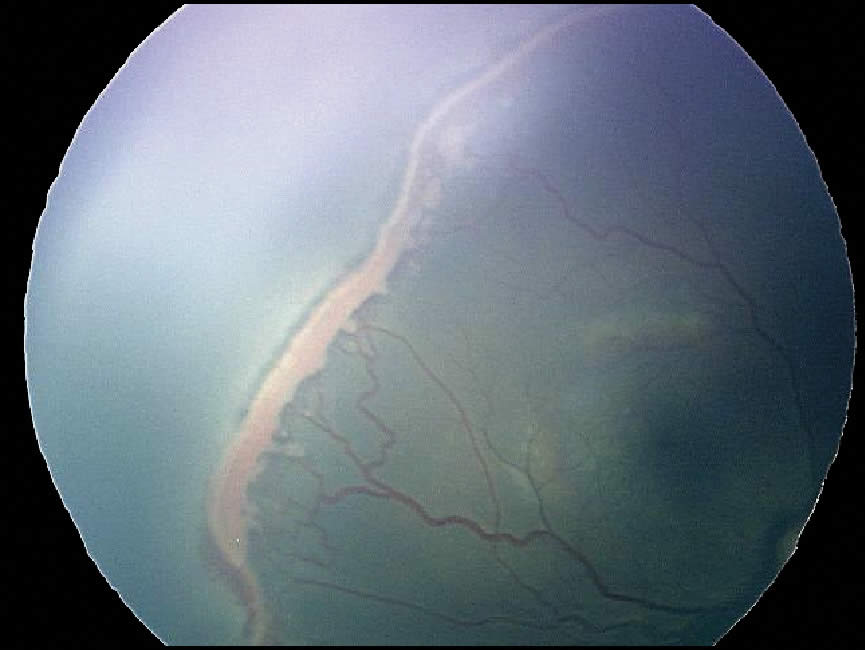
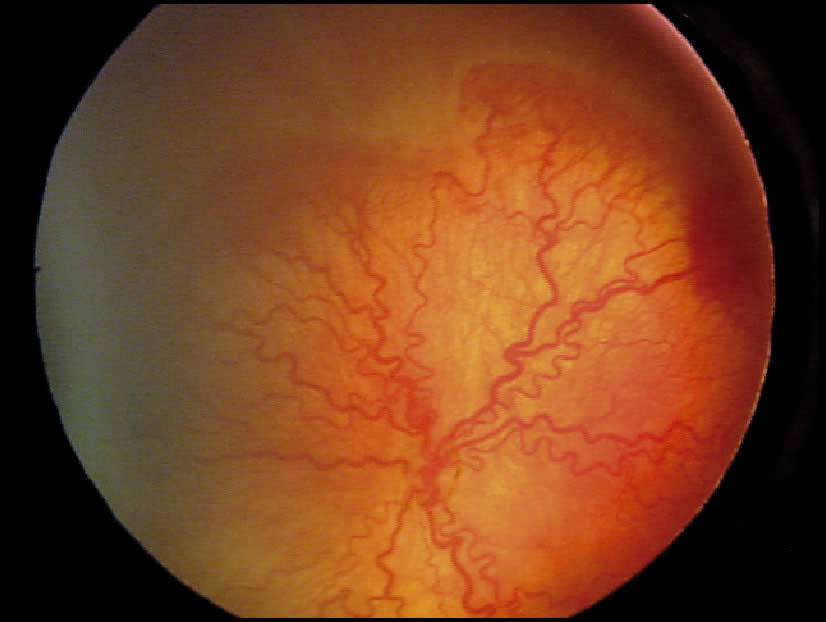
Retinal detachment
A retinal detachment occurs when the retina’s neurosensory layer and pigment epithelial layers separate. There are three types of retinal detachments:
Rhegmatogenous
This is the most common type and occurs when there is a break in the sensory layer of the retina, and liquefied vitreous seeps underneath, causing the two layers of the retina to separate.
Tractional
The second most common type occurs when strands of vitreous or scar tissue create traction on the retina, pulling it loose.
Exudative
This results from an accumulation of fluid under an intact neurosensory retina. This usually occurs in conjunction with another disease, e.g posterior scleritis, choroidal inflammatory conditions and neoplasms.
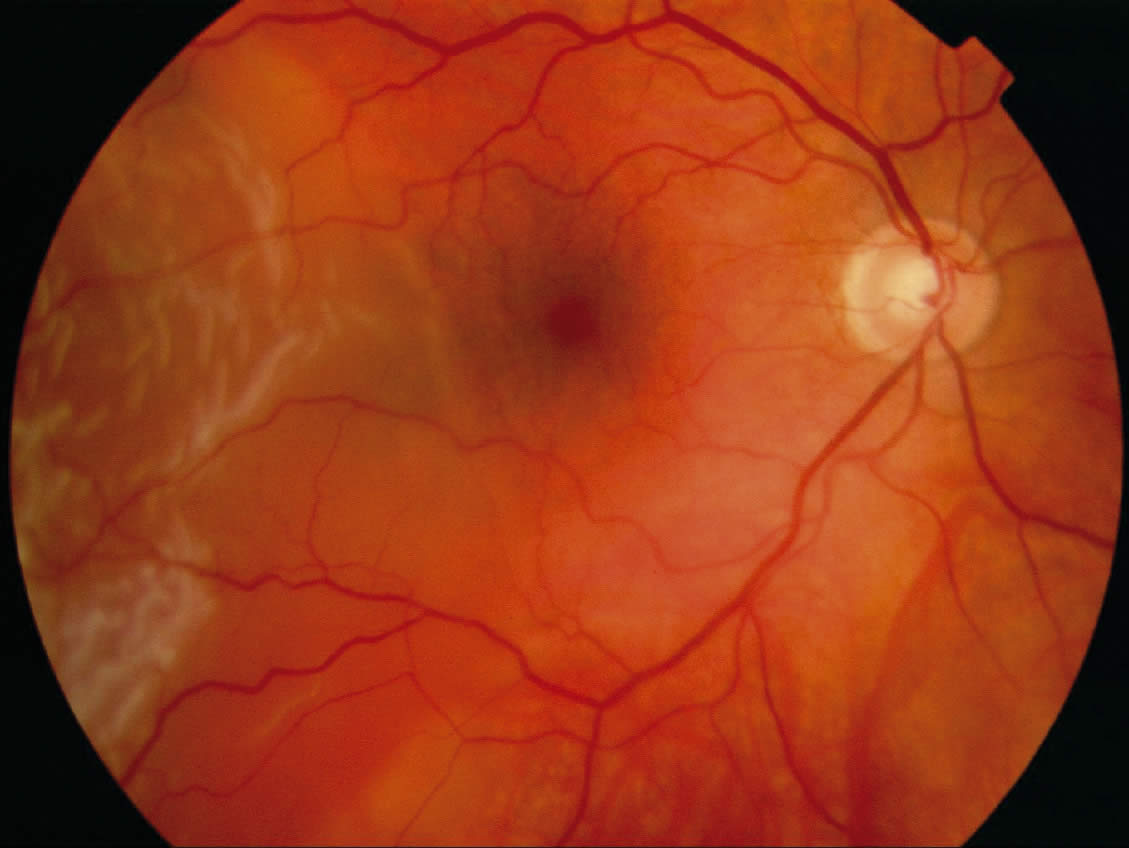
Retinoblastoma
Retinoblastoma is a primary malignant intraocular neoplasm that arises from immature retinoblasts within the developing retina. It is the most common primary intraocular malignancy of childhood. Most cases occur in children younger than six years of age.
The most common presenting symptoms of retinoblastoma are leukocoria (a white pupil), in the tumourcontaining eye or eyes, strabismus or symptomatic or asymptomatic visual loss. Retinoblastoma can be hereditary.
Retinal dystrophies – Retinitis pigmentosa (RP)
A group of hereditary retinal conditions that cause degeneration of the retina.
Retinal cells are among the most specialised cells in the human body and depend on a number of unique genes to create vision. A disease-causing mutation in any one of these genes can lead to vision loss. RP results from a large and as yet unknown number of gene defects, of which around a hundred have been found so far. RP can be passed to succeeding generations by one of three genetic inheritance patterns: autosomal dominant, autosomal recessive, or X-linked inheritance. RP causes the degeneration of photoreceptor cells from the outer edges of the retina, causing a progressive loss of peripheral vision, night blindness and reduced or absent electroretinogram (ERG) recordings.